Drug-tolerance
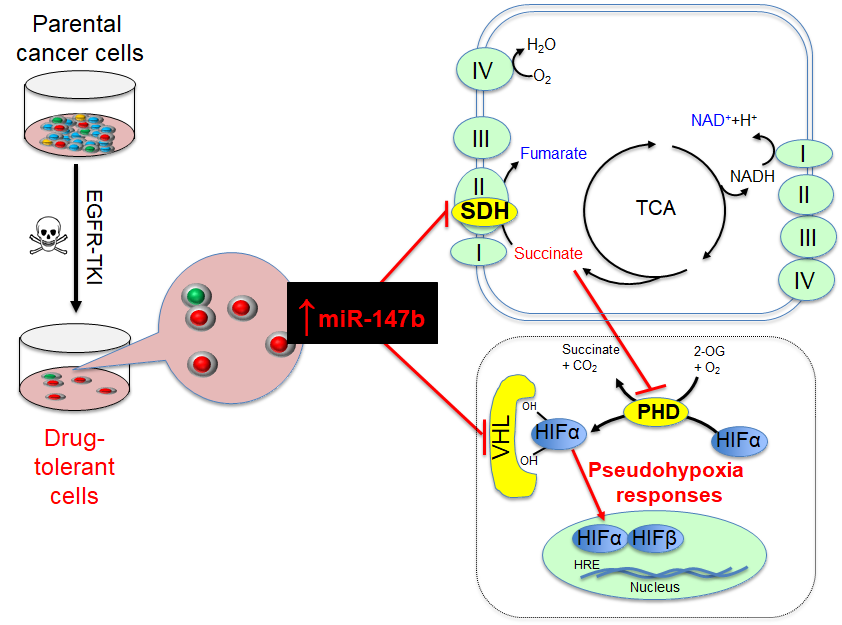
Relapse of disease following conventional treatments remains one of the central problems in cancer management, including in EGFR-based therapy. Tumor cells overcome EGFR-targeted treatment through the acquisition of mutations that disrupt drug binding by EGFR and activate signaling through other protein tyrosine kinase pathways. For example, a majority of tumors from patients with EGFR-mutant non-small-cell lung cancer (NSCLC) were shown to have acquired resistance-conferring mutations when patients were treated with EGFR tyrosine kinase inhibitors (TKIs), including EGFRT790M on gefitinib treatment and EGFRC797S on combination erlotinib and osimertinib therapy. Recently, it shows that EGFRT790M-positive drug-resistant cells can emerge from EGFRT790M-negative drug-tolerant cells that survive initial drug treatment. Thus, targeting drug-tolerant cells might be a new strategy to block drug resistance. With the success in applying osimertinib in first-line treatment of EGFRT790M-positive NSCLC, it is crucial to identify the changes driving drug tolerance. We have examined whole transcriptome and metabolomics profiles in non-small lung cancer cells during their survival/tolerance to targeted therapies and discovered that the tricarboxylic acid (TCA) pathway, irrespective of oxygen levels, confers anticancer drug tolerance. Our results also reveal that microRNA-147b, targeting von-Hippel Lindau (VHL) and succinate dehydrogenase (SDH), is critical to tolerance-mediated tumor relapse. Briefly, our extensive findings reveal that a subpopulation of tumor cells adopt a tolerance strategy to defend against anticancer treatments by altering microRNA-147b-dependent dysregulation of mitochondrial respiration and pseudohypoxia responses. The reduced oxidized nicotinamide adenine dinucleotide (NAD+) and glutathione (GSH) coordinate with repressed SDH and VHL for activation of the pseudohypoxia responses and drug-tolerance. Given that tolerance precedes and promotes the emergence of mutational resistance in anticancer, anti-aging, anti-bacterial, and anti-viral treatments, as well as the fact that microRNA-147b-mediated dysfunctional TCA cycle and activated pseudohypoxia signaling pathways coordinately drive drug-tolerance, we believe that our interdisciplinary findings will appeal to pharmaceutical scientists, biologists, and clinicians in fields of cancer, aging and infectious diseases.
Lung Precancer Development
Tumor initiates from precancerous lesions and advances towards invasive carcinoma. Adenocarcinoma (AC) is the most common subtype in lung cancer. The precursors of lung AC include atypical adenomatous hyperplasia (AAH) and adenocarcinoma in situ (AIS). The most recent genomic profile for lung precancer has shown that the precancerous lesions share the same driving mutations to paired advanced AC, including KRAS, BRAF, and EGFR. These precancerous lesions either regress or persist long periods before progression into invasive carcinoma, suggesting alternative signaling pathways besides KRAS, BRAF, and EGFR signaling pathways regulate lung precancerous development. However, it is unclear about the molecular mechanism of precancer initiation and progression into invasive AC. The lab is utilizing comprehensive approaches including genetics, transcriptomics, and metabolomics as well as cutting-edge technology including 3D spheroid/organoid and transgenic mouse models to study which factors play roles in precancer development.
Lung Cancer microRNAs
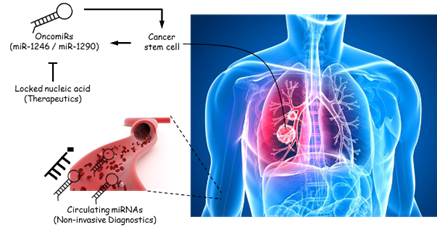
This study brings together three important fields: tumor-initiating cell (TIC), circulating cell-free microRNA (miRNA) as a biomarker for therapy-response, and miRNA as a therapeutic target. In recent years, there have been tremendous interests in drugging TICs as a novel therapeutic strategy and much effort has been devoted to identifying targets unique to TICs. One approach we demonstrate here is through the inhibition of TIC-specific miRNAs that are attractive therapeutic targets. In this study, we focus primarily on the use and interrogation of patient-derived clinical materials from non-small cell lung cancer patients to identify and functionalize key miRNAs that are restricted to lung TICs. Our data clearly indicate that miR-1246 and miR-1290, both of which have not been previously implicated in TIC function and, in fact, are not extensively examined in the context of cancer, have clear clinical implications when found altered in tumors. For the first time, we were able to track changes in circulating miRNA levels during on-going treatment and correlate this information with the clinical response of patients to therapy, clearly demonstrating the utility of this method as a diagnostic and prognostic strategy, especially in detecting metastasis not easily seen on computed tomography (CT) scan. We can rigorously dissect the function of these miRNAs, particularly in promoting tumor initiation and metastasis, as well as mechanistically define one of their major targets, MT1G (a metallothionein protein), which is common to both miRNA species. From a therapeutic angle, we are successful in employing locked nucleic acid technology to specifically silence the miRNAs and demonstrating a direct impact on the inhibition of tumor initiation and the arrest of growth for pre-established tumors. Together, these results clearly place miR-1246 and miR-1290 as viable therapeutic targets, which have the potential to favorably impact clinical outcomes in lung cancer patients. I served as the project leader in these studies.
Lung Cancer Metabolism
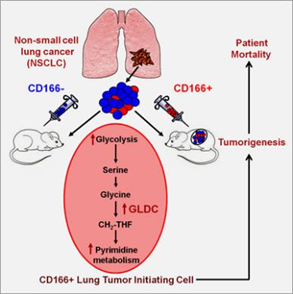
We showed that the surface marker CD166+ consistently selects and enriches by over a hundred-fold tumor-initiating cells (TICs) in vivo and in vitro in a broad range of non-small cell lung cancers, the leading cause of cancer mortality worldwide. By whole transcriptome analysis of the TICs, we showed that TICs overexpressed the metabolomic enzyme Glycine Decarboxylase (GLDC) and several other enzymes in the glycine/serine/threonine metabolism pathway. GLDC is necessary for lung cancer growth since knockdown of GLDC results in significantly lower clonal growth in vitro and smaller tumors in vivo. Surprisingly, overexpression of GLDC alone transforms fibroblasts that form tumors in vivo. Metabolomic studies suggest that overexpression of GLDC resulted in enrichment in glycine/serine/threonine metabolism, pyrimidine metabolism, and glycolysis/pyruvate metabolism pathway and increase in cell proliferation. Clinically, GLDC overexpression is significantly associated with higher mortality in lung cancer patients. In a survey of other cancers, we find that GLDC is overexpressed in many other types of major cancers, and knockdown of GLDC in other cancers also significantly reduces tumor growth in vivo. Altogether, our results directly link a novel enzyme in glycine metabolism to the proliferation and survival of cancer cells. These new findings have a significant impact on the prognostication and treatment of cancers. Our findings add to the increasing awareness of the fundamental difference between the metabolism of cancer and normal cells and to the recent discoveries of druggable enzymes in metabolic pathways.